Recall(So far in this series)
Hallmark #1: cancer generates its own growth signals. Hallmark #2: cancer ignores signals telling it to stop. Hallmark #3 completes the picture — even when a cell is damaged beyond repair, cancer finds ways to stay alive anyway.
Every multicellular organism runs a quiet program of self-destruction. Cells that are damaged, infected, mislocated, or simply no longer needed are eliminated — not by being killed from outside, but by activating their own death machinery from within. This process is called apoptosis, and it is as fundamental to healthy tissue as cell division itself.
Cancer's third move is to disable it.
What apoptosis actually is
Apoptosis is not necrosis. When a cell dies by necrosis — from trauma, toxins, or oxygen deprivation — it ruptures, spilling its contents and triggering inflammation. Apoptosis is the opposite: an orderly, contained dismantling. The cell shrinks, its DNA is cleaved into fragments, its membrane buds into tidy vesicles, and neighboring cells or immune phagocytes clear the debris without any inflammatory signal.
Definition(Apoptosis)
A form of programmed cell death characterized by cell shrinkage, chromatin condensation, DNA fragmentation, and membrane blebbing. Executed by a family of proteases called caspases. Apoptosis is non-inflammatory — the cell is packaged and cleared without damaging surrounding tissue. Distinct from necrosis (traumatic, inflammatory) and autophagy (self-digestion for survival under nutrient stress).
The machinery of apoptosis is built around caspases — a family of proteases that exist in cells as inactive precursors and, once activated, cleave hundreds of cellular proteins in a coordinated cascade. Activation of caspase-3 (the "executioner" caspase) is essentially the point of no return: the cell is committed to dying.
The question is how caspases get activated. There are two main routes.
The two paths to cell death
The intrinsic pathway — triggered from within
The intrinsic pathway is the one most relevant to cancer. It's activated by internal stress signals: DNA damage, oxidative stress, oncogene overactivation, loss of survival signals.
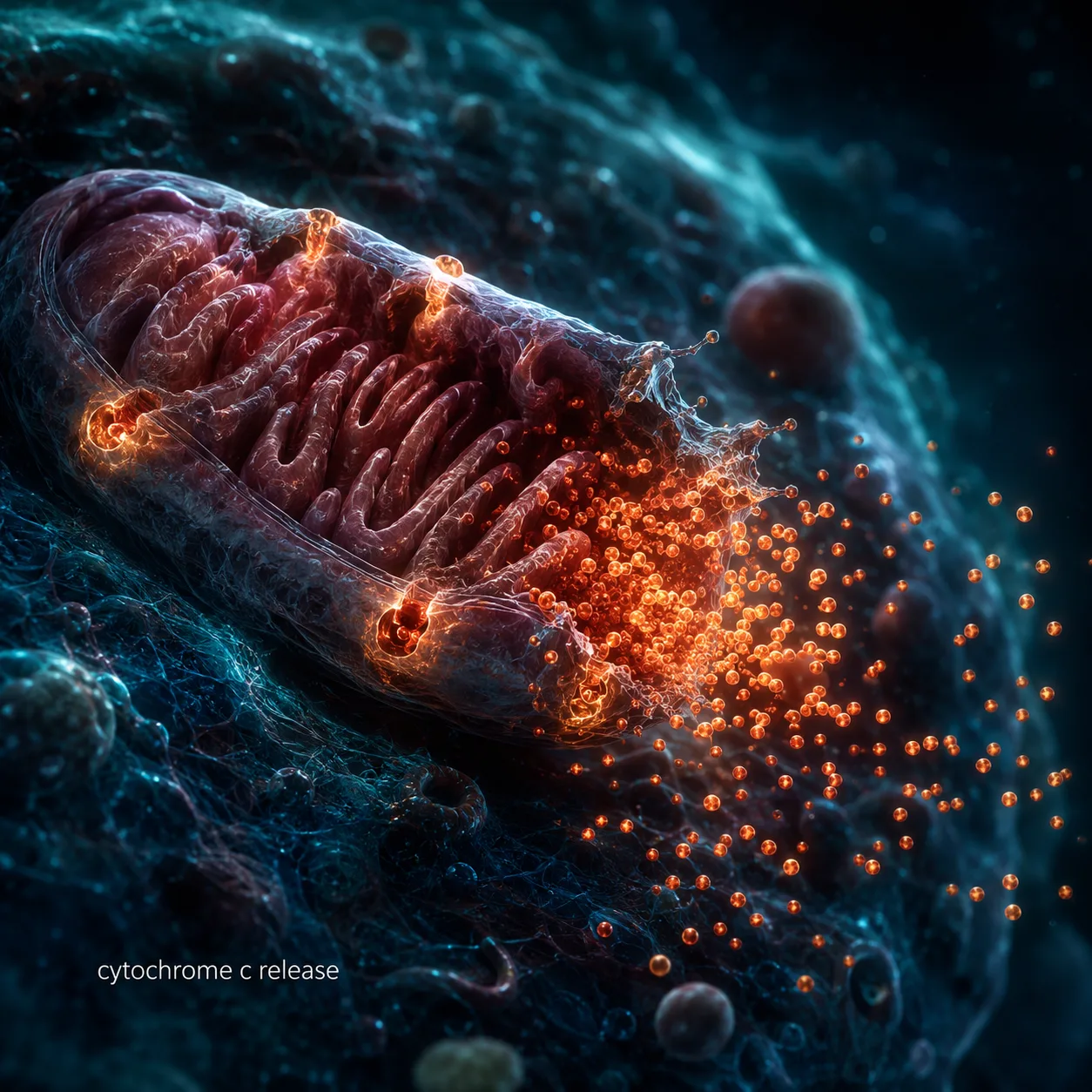
The decision point is the mitochondria. Pro-apoptotic proteins (BAX, BAK) and anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1) are in constant competition at the mitochondrial membrane. When pro-apoptotic signals dominate, BAX and BAK form pores in the mitochondrial outer membrane, releasing cytochrome c into the cytoplasm. Cytochrome c then assembles a structure called the apoptosome, which activates caspase-9, which activates caspase-3.
Definition(BCL-2 family)
A family of proteins that regulate apoptosis by controlling mitochondrial membrane permeability. Divided into pro-apoptotic members (BAX, BAK, BIM, PUMA, NOXA, BID) and anti-apoptotic members (BCL-2, BCL-XL, MCL-1, BCL-W). The balance between these two groups determines whether a cell lives or dies in response to stress.
Cancer tilts this balance toward survival in several ways:
BCL-2 overexpression was the first mechanism identified. In follicular lymphoma, a chromosomal translocation (t14;18) places the BCL-2 gene under the control of an immunoglobulin promoter — a promoter that's constitutively active in B cells. The result is BCL-2 expressed at high levels in every cell, sequestering pro-apoptotic proteins and preventing BAX/BAK pore formation even under stress.
MCL-1 amplification has emerged as a major resistance mechanism across many cancer types. MCL-1 is particularly potent because it has a short half-life — it turns over rapidly — which means cancer cells often become dependent on continuous MCL-1 expression to suppress apoptosis.
BIM deletion or silencing removes one of the key activators of BAX/BAK. Without BIM, even properly functioning BCL-2 family balance is harder to tip toward death.
Example(Why venetoclax works — and why resistance happens)
Venetoclax is a BCL-2 inhibitor approved for CLL and AML. It works by mimicking BH3-only proteins — it binds the groove in BCL-2 that normally sequesters BAX and BAK activators, freeing them to trigger apoptosis. Resistance frequently emerges through MCL-1 upregulation: the tumor switches its dependence from BCL-2 to MCL-1, which venetoclax doesn't target. This is why MCL-1 inhibitors are now in clinical trials as combination partners.
The extrinsic pathway — triggered from outside
The extrinsic pathway is activated by death ligands — signals from the immune system or neighboring cells that tell a cell it needs to die. The key players are FasL (Fas ligand) binding to Fas receptor, and TRAIL binding to death receptors DR4/DR5.
When a death ligand binds its receptor, it assembles a death-inducing signaling complex (DISC) that directly activates caspase-8, which then activates caspase-3.
Cancer evades this pathway by:
- Downregulating death receptors (less Fas or DR5 on the cell surface)
- Upregulating FLIP, a decoy protein that competes with caspase-8 for DISC binding without activating it
- Secreting decoy receptors that sequester TRAIL before it reaches the cell surface
The extrinsic pathway is particularly relevant to immune-mediated killing — cytotoxic T cells and NK cells use FasL and TRAIL to eliminate target cells. A tumor that downregulates death receptors is partly shielding itself from immune attack (which connects to hallmark #8).
p53: the failsafe that fails in most cancers
p53 appears here again, which illustrates why its loss is so catastrophic. In hallmark #2, p53 enforces cell cycle arrest in response to damage. But p53 also drives apoptosis — it transcriptionally activates PUMA and NOXA (BH3-only pro-apoptotic proteins) and BAX itself.
A cell without p53 doesn't just fail to arrest when damaged. It also fails to die when it should. The surveillance system is gone in both directions.
Intuition(Why most chemotherapy depends on functional apoptosis)
Classic cytotoxic chemotherapy — platinum compounds, taxanes, anthracyclines — kills cancer cells primarily by inducing enough DNA damage or mitotic disruption to trigger apoptosis. Tumors with disabled apoptotic machinery (BCL-2 overexpression, p53 loss, caspase mutations) are frequently resistant to these agents. The drug causes damage; the cell just doesn't die from it. This is one of the root causes of chemotherapy resistance.
Other forms of programmed death
Apoptosis gets most of the attention, but it's not the only cell death program cancer has to contend with:
Autophagy is a survival mechanism where the cell digests its own organelles and proteins for energy under nutrient stress. In cancer it plays a complex dual role — it can promote survival under hypoxia and nutrient deprivation (helping established tumors survive), but excessive autophagy can also lead to cell death. The relationship between autophagy and cancer is context-dependent and still being worked out.
Anoikis is apoptosis triggered by loss of attachment to the extracellular matrix. Normal epithelial cells require contact with their substrate to survive — if they detach, they die. Cancer cells that metastasize must evade anoikis to survive in circulation and colonize distant sites. This connects directly to hallmark #6 (invasion and metastasis).
Necroptosis is a form of programmed necrosis — inflammatory cell death mediated by RIPK3 and MLKL. Some evidence suggests tumors downregulate necroptosis machinery to avoid triggering immune responses that might attract immune surveillance.
Summary(Summary)
Hallmark #3 is about survival in the face of signals that should be lethal. Cancer resists apoptosis primarily by overexpressing anti-apoptotic BCL-2 family members (BCL-2, MCL-1), losing pro-apoptotic activators (BIM, PUMA), downregulating death receptors (Fas, DR4/DR5), and losing p53 — which drives apoptosis as well as cell cycle arrest. The clinical consequence is that tumors with robust apoptosis resistance are frequently resistant to conventional chemotherapy, since most cytotoxics kill through apoptosis induction. Therapies like venetoclax attempt to directly re-engage the pathway by blocking BCL-2, but resistance through pathway switching (MCL-1 upregulation) is common.